
TRUNG TÂM XÉT NGHIỆM ADN LAB SÀI GÒN
234
Chủ Nhật, 30/08/2024, 06:40 (GMT+7)
Thalassemia là căn bệnh di truyền khá phổ biến hiện nay. Hằng năm, tại Việt Nam tiếp nhận và điều trị khoảng 1 nghìn người mắc phải căn bệnh di truyền này. Để giúp các bạn hiểu rõ căn bệnh Thalassemia là gì, các phát hiện cũng như các phương pháp điều trị. Chúng tôi sẽ mang đến những thông tin hữu ích qua bài viết sau đây.
Là căn bệnh phổ biến hiện nay, tuy nhiên không nhiều người biết đến bệnh Thalassemia. Vậy căn bệnh Thalassemia là gì? Cách phát hiện như thế nào qua từng mức độ như thế nào?
Bệnh thalassemia là một rối loạn di truyền gen lặn trên nhiễm sắc thể thường gây tan máu, thiếu máu và ứ sắt.
Những người mắc bệnh thalassemia không sản xuất đủ lượng huyết sắc tố khỏe mạnh, khiến các tế bào máu của họ nhỏ và nhợt nhạt. Hemoglobin là một loại protein được tìm thấy trong các tế bào hồng cầu mang oxy từ phổi đến phần còn lại của cơ thể.
Những người sinh ra mắc bệnh thalassemia không thể vận chuyển oxy khắp cơ thể một cách bình thường. Tùy thuộc vào loại bệnh thalassemia mà họ mắc phải, họ có thể cần được truyền máu thường xuyên để sống sót.
Thalassemia được chia thành 2 loại chính là:
Dù bệnh nhân mắc loại nào thì cũng đều tiến triển và biểu hiện ở 3 mức độ sau đây:
Đối với người mắc bệnh Thalassemia ở mức độ này rất khó phát hiện vì bệnh hầu như không có biểu hiện khác biệt nào về mặt lâm sàng. Tuy nhiên, khi cơ thể cần nhiều máu, nhất là đối với phụ nữ trong các thời kỳ kinh nguyệt hay mang thai. Lúc này bệnh sẽ thể hiện ra với các triệu chứng như mệt mỏi hay da xanh xao và việc suy giảm hồng cầu.
Người bệnh có thể được phát hiện sớm hơn so với mức độ nhẹ. Người bệnh thalassemia thể trung gian sẽ cần được truyền máu trong giai đoạn từ 4 đến 6 tuổi. Dù không quá nguy hiểm tuy nhiên nếu không được can thiệp và đưa ra phương pháp điều trị kịp thời, bệnh sẽ diễn biến nặng và gây ra các biên chứng như gan to, sạm da hay sỏi mật. Nếu được phát hiện sớm và bổ sung máu thường xuyên cơ thể sẽ có thể phát triển bình thường.
Đối với mức độ này, người bệnh có thể được phát hiện nay sau khi ra đời với các biểu hiện như xanh xao, da vàng và chậm phát triển về thể chất, thường xuyên gặp các vấn đề về rối loạn tiêu hóa và sốt. Bệnh được biểu hiện rõ nhất khi trẻ từ 4 đến 6 tháng tuổi.
Nếu không được can thiệp kịp thời, bệnh Thalassemia ở mức độ nặng sẽ để lại nhiều di chứng khá nghiêm trọng như biến dạng các bộ phận xương trong cơ thể, các bệnh lý về xương hay dậy thì muộn.
Bệnh thalassemia xảy ra khi ai đó thừa hưởng đột biến gen (là các ‘lỗi chính tả’ trong trình tự ADN ở gen) từ một hoặc cả hai cha mẹ. Những đột biến gen này khiến cơ thể mất tế bào hồng cầu nhanh hơn bình thường và dẫn đến lượng huyết sắc tố ít hơn.
Có nhiều loại thalassemia khác nhau. Loại bệnh thalassemia mắc phải sẽ phụ thuộc vào loại đột biến gen mà họ thừa hưởng.
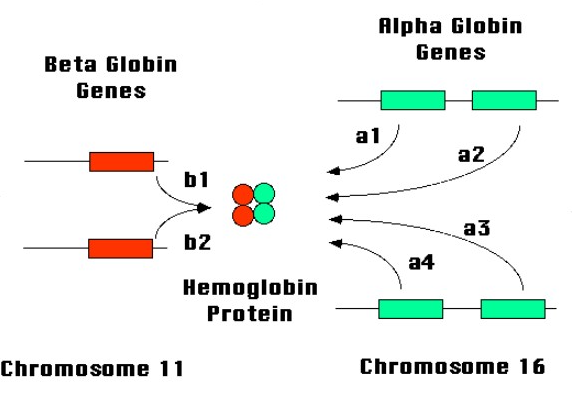
Điều này xảy ra do có vấn đề ở 1 hoặc nhiều hơn trong số 4 gen định vị trên nhiễm sắc thể số 16 được gọi là gen alpha globin.
Bình thường có bốn alen alpha (hai trên mỗi cặp nhiễm sắc thể) vì gen alpha được nhân đôi. Phân loại bệnh dựa trên số lượng gen bị mất:
Những người chỉ thừa hưởng 1 gen có thể không có bất kỳ triệu chứng nào nhưng họ vẫn có thể truyền bệnh cho con cái. 2 gen gây ra các triệu chứng nhẹ, 3 gen (gọi là bệnh Hemoglobin H) gây ra các triệu chứng nghiêm trọng hơn và những em bé thừa hưởng 4 gen (được gọi là alpha thalassemia thể nặng hoặc phù thai) thường bị bệnh nặng và không sống được lâu sau khi sinh ra.
Alpha thalassemia phổ biến hơn ở những người gốc Đông Nam Á, Nam Trung Quốc, Trung Đông, Ấn Độ, Châu Phi hoặc Địa Trung Hải.
Beta thalassemia là kết quả của sự giảm sản xuất chuỗi beta do đột biến gen hoặc mất gen quy định chuỗi beta, dẫn đến hư hại sản xuất Hemoglobin (Hb) A
Đột biến xảy ra ở 1 hoặc 2 gen định vị trên nhiễm sắc thể số 11 gọi là gen beta globin.
Các đột biến hoặc mất gen có thể dẫn đến mất một phần (beta +) hoặc mất hoàn toàn (beta 0) của chức năng beta globin. Có hai gen beta globin, và bệnh nhân có thể đột biến dị hợp tử, đồng hợp tử, hoặc phức hợp dị hợp tử.
Ngoài ra, bệnh nhân có thể là dị hợp tử hoặc đồng hợp tử cho những bất thường ở 2 gen globin khác nhau (ví dụ beta và delta).
Beta-delta-thalassemia là một hình thức ít phổ biến hơn, trong đó sản xuất cả chuỗi delta cũng như chuỗi beta bị suy giảm. Những đột biến này có thể là dị hợp tử hoặc đồng hợp tử.
Có hàng trăm loại đột biến có thể xảy ra và các triệu chứng tùy thuộc vào loại đột biến mà một người mắc phải. Những người chỉ có 1 gen bị lỗi thường có triệu chứng tối thiểu nhưng họ có thể truyền bệnh thalassemia cho con cái. Những người có 2 gen bị lỗi (gọi là thalassemia thể nặng) thường có những triệu chứng nguy hiểm đến tính mạng. Một số người có 2 gen bất thường vẫn có thể có các triệu chứng nhẹ hơn và chỉ cần truyền máu thường xuyên.
Beta thalassemia thường ảnh hưởng đến người gốc Địa Trung Hải, châu Á hoặc châu Phi.
Các đặc điểm lâm sàng của thalassemia giống nhau nhưng khác nhau về mức độ nghiêm trọng tùy thuộc vào lượng Hemoglobin bình thường.
Bệnh nhân khiếm khuyết một alpha + alen (alpha/alpha;alpha/–) có lâm sàng bình thường (người mang gen).
Bệnh nhân có dị hợp tử với khiếm khuyết ở 2 trong 4 gen như hai alen alpha + (alpha/–;alpha/–) hoặc một alen alpha 0 (alpha/alpha;–/–) có xu hướng phát triển bệnh thiếu máu hồng cầu nhỏ từ nhẹ đến trung bình nhưng không có triệu chứng. Những bệnh nhân này mang bệnh alpha thalassemia.
Khiếm khuyết ở 3 trong số 4 gen gây ra bởi sự đồng nhất của cả alpha + và alpha 0 (alpha/–;–/–) làm suy giảm nghiêm trọng việc sản xuất chuỗi alpha. Điều này dẫn đến sự hình thành các tetramer của các chuỗi beta dư thừa được gọi là Hb H, hoặc ở trẻ nhỏ hình thành chuỗi gamma được gọi là Hb Bart. Bệnh nhân Hb H thường mắc thiếu máu tan máu có triệu chứng và lách to.
Khiếm khuyết ở tất cả 4 gen thông qua hai alen alpha 0 (–/–;–/–) là một tình trạng nguy hiểm, gây phù thai trong tử cung, bởi vì hemoglobin thiếu chuỗi alpha không thể vận chuyển oxy.
Trong beta-thalassemia, các kiểu hình lâm sàng được phân thành 3 nhóm dựa trên mức độ sản xuất beta globin bị suy giảm:
Beta-thalassemia thể nhẹ (trait) xảy ra ở những người có dị hợp tử (beta/beta + hoặc beta/beta 0), những người này thường không có triệu chứng kèm bệnh thiếu máu hồng cầu nhỏ từ nhẹ đến trung bình. Kiểu hình này cũng có thể xảy ra trong các trường hợp nhẹ của beta +/beta +.
Beta-thalassemia thể trung gian là một bệnh cảnh lâm sàng biến đổi trung gian giữa thalassemia thể nặng hay nhẹ, do di truyền 2 alen beta thalassemia (beta +/beta 0 hoặc các trường hợp nặng beta +/beta +).
Beta-thalassemia nặng (hoặc thiếu máu Cooley) là dạng đồng hợp tử (beta 0/beta 0) hoặc dị hợp tử kép (chứa một alen beta 0/beta +), là kết quả khi thiếu hụt globin beta nghiêm trọng. Những bệnh nhân này bị thiếu máu trầm trọng và tủy xương tăng sản mạnh. Beta-thalassemia nặng biểu hiện từ 1-2 tuổi với các triệu chứng thiếu máu trầm trọng và quá tải sát. Bệnh nhân bị vàng da, loét chân và sỏi mật (như trong bệnh hồng cầu liềm). Thường có lách to, thường rất lớn. Tăng phá hủy các hồng cầu bình thường được truyền tại lách. Tăng hoạt động tủy xương để sinh máu làm bè xương sọ, xương gò má. Gãy bệnh lý các xương dài, giảm phát triển chiều cao, chậm dậy thì.
Quá tải sắt, nhiễm sắt trong cơ tim có thể gây suy tim. Gan nhiễm sắt, dẫn đến suy giảm chức năng và xơ gan. Cần thải sắt.
Nhìn chung, cho dù là dạng nào thì người bị bệnh thalassemia có thể có những biểu hiện:
Nếu không được điều trị, bệnh thalassemia có thể dẫn đến suy tim và nhiễm trùng do ứ sắt.
Các triệu chứng có thể xuất hiện ở trẻ sơ sinh và trẻ nhỏ nếu tình trạng bệnh nghiêm trọng, thường là trong 2 năm đầu đời.
Khi đã biết bệnh Thalassemia là gì? Mọi người đều cần quan tâm đến phương pháp điều trị của bệnh. Hiện nay, dù bệnh ở mức độ nào thì cũng thường được sử dụng các phương pháp điều trị chính sau.
Đây là phương pháp điều trị giúp loại bỏ lượng sắt dư thừa bằng cách uống hoặc tiêm các tế đơn vị hồng cầu lắng. Phương pháp này sẽ được thực hiện điều trị cả đời để giúp bệnh nhân không bị dư lượng sắt trong cơ thể vì chúng có thể dẫn đến nhiều biến chứng xấu.
Đối với phương pháp này, bệnh nhân mắc bệnh Thalassemia sẽ được truyền hồng cầu trực tiếp vào cơ thể. Mức truyền phổ biến là 7g/dl nếu mức độ bệnh nhẹ và tăng lên nếu cơ thể gặp phải tình trạng biến dạng xương. Người bệnh thực hiện phương pháp điều trị này cần được theo dõi thường xuyên tại bệnh viện để xử lý các vấn để trong quá trình điều trị.

Phương pháp này ít khi được sử dụng trong việc điều trị bệnh Thalassemia. Chỉ khi quá trình cung cấp máu cho cơ thể không đạt được hiệu quả, biến chứng lá lách to ra sẽ gây ra đau, ảnh hưởng đến sức khỏe, lúc này việc cắt bỏ bớt lá lách sẽ cần thiết và được thực hiện.
Đây là phương pháp sử dụng đối với những người mắc bệnh ở mức độ nặng và có thể điều trị dứt điểm được bệnh Thalassemia. Hiện nay, tại Việt Nam có nhiều bệnh viện lớn đã thực hiện thành công trong việc điều trị bệnh bằng phương pháp này.
Điểm khó khăn của phương pháp này là việc tìm kiếm được tế bào gốc phù hợp. Bên cạnh đó, việc ghép tế bào gốc cũng dễ dẫn đến việc lây nhiễm các căn bệnh như viêm gan hay lao phổi,…
Rất nhiều người chưa biết căn bệnh Thalassemia là gì khiến họ chủ quan và không thường xuyên kiểm tra sức khỏe. Điều này sẽ ảnh hưởng nhiều đến quá trình phát hiện và điều trị bệnh.
Đối với bệnh nhân khi thấy cơ thể có các biểu hiện như mệt mỏi, cơ thể yếu, da vàng, xương bị biến dạng hay việc chậm phát triển thì nên đến các trung tâm y tế để thực hiện xét nghiệm máu. Bên cạnh đó, đối với các gia đình từng có người mắc Thalassemia cũng nên đi kiểm tra để phát hiện kịp thời.
Thực hiện các xét nghiệm nhằm kiểm tra bệnh Thalassemia kịp thời
Hiện nay, Bệnh Thalassemia có thể được phát hiện khi thực hiện các xét nghiệm máu sau:
Những người mắc bệnh thalassemia có thể phát triển các vấn đề khác bao gồm:
Hy vọng những thông tin của bài viết đã giúp các bạn biết được bệnh Thalassemia là gì? Cách phát hiện và các phương pháp điều trị phổ biến hiện nay. Hãy sớm thực hiện các xét nghiệm tại các cơ sở ý tế chất lượng để phát hiện và điều trị kịp thời căn bệnh nguy hiểm này.




