
TRUNG TÂM XÉT NGHIỆM ADN LAB SÀI GÒN
Nguyên lý giải trình tự thế hệ mới
614
Thứ Hai, 09/11/2023, 03:03 (GMT+7)

Kể từ khi 2 nhà khoa học Watson và Crick khám phá ra cấu trúc chuỗi xoắn kép của phân tử ADN, các nhà nghiên cứu đã phát triển các phương pháp và công nghệ hỗ trợ xác định trình tự axit nucleic trong các mẫu sinh học. Khả năng giải trình tự ADN và ARN một cách chính xác của các hệ thống giải trình tự đã có tác động lớn đến nhiều lĩnh vực nghiên cứu. Bài viết này thảo luận về nguyên lý giải trình tự thế hệ mới (Next-Generation Sequencing, NGS), những tiến bộ trong công nghệ và các ứng dụng của NGS.
Giải trình tự thế hệ mới là gì?
Cấu trúc của ADN được xác định vào năm 1953 bởi Watson và Crick dựa trên phương pháp tinh thể học ADN cơ bản và nghiên cứu nhiễu xạ tia X của Rosalind Franklin.
Tuy nhiên, phân tử đầu tiên được giải trình tự thực chất là ARN – tRNA – vào năm 1965 bởi Robert Holley và ARN của thực khuẩn thể MS2 sau này.
Sau đó, nhiều nhóm nghiên cứu khác nhau bắt đầu áp dụng các phương pháp này để giải trình tự ADN với một bước đột phá vào năm 1977 bởi Frederick Sanger và các đồng nghiệp, phát triển phương pháp kết thúc chuỗi.
Đến năm 1986, phương pháp giải trình tự ADN tự động đầu tiên đã được phát triển. Đây là sự khởi đầu của một kỷ nguyên vàng cho sự phát triển và hoàn thiện các nền tảng giải trình tự, bao gồm cả máy giải trình tự ADN mao quản quan trọng.
Phương pháp kết thúc chuỗi, còn được gọi là giải trình tự Sanger, sử dụng trình tự ADN quan tâm làm mẫu cho PCR để bổ sung các nucleotide đã biến đổi, được gọi là dideoxyribonucleotide (ddNTPs), vào chuỗi ADN trong bước mở rộng.
- Khi DNA polymerase kết hợp với ddNTP, quá trình mở rộng sẽ ngừng dẫn đến việc tạo ra nhiều bản sao của chuỗi ADN có đủ độ dài trải dài trên đoạn được khuếch đại.
- Các oligonucleotide kết thúc chuỗi này sau đó được phân tách kích thước bằng cách sử dụng điện di trên gel trong các phương pháp trước đây hoặc các ống mao quản trong các máy giải trình tự mao quản tự động sau này và trình tự ADN được xác định.
Với những tiến bộ công nghệ đột phá này, dự án bộ gen người đã được hoàn thành vào năm 2003.
Vào năm 2005, nền tảng NGS thương mại đầu tiên, hay còn gọi là giải trình tự gen thế hệ thứ hai (2G), đã được giới thiệu, có khả năng khuếch đại hàng triệu bản sao của một đoạn ADN cụ thể theo cách song song ồ ạt, trái ngược với trình tự Sanger.
Các nguyên tắc chính đằng sau công nghệ giải trình tự Sanger và NGS thế hệ thứ 2 có một số điểm tương đồng.
Trong NGS thế hệ thứ 2, vật chất di truyền (ADN hoặc ARN) được phân mảnh, sau đó các oligonucleotide của các trình tự đã biết được gắn vào đó, thông qua một bước được gọi là nối bộ chuyển đổi, cho phép các đoạn tương tác với hệ thống giải trình tự đã chọn. Các cơ sở của mỗi mảnh sau đó được xác định bằng các tín hiệu phát ra của chúng.
Sự khác biệt chính giữa giải trình tự Sanger và NGS thế hệ thứ 2 bắt nguồn từ khối lượng giải trình tự, trong đó NGS cho phép xử lý song song hàng triệu phản ứng, mang lại hiệu suất cao, độ nhạy, tốc độ cao hơn và giảm chi phí.
Rất nhiều dự án giải trình tự bộ gen từng mất nhiều năm sử dụng phương pháp giải trình tự Sanger giờ đây có thể được hoàn thành trong vòng vài giờ bằng NGS.
Có hai cách tiếp cận chính trong công nghệ NGS, giải trình tự đọc ngắn và đọc dài, mỗi phương pháp đều có những ưu điểm và hạn chế riêng (Bảng 1).
Phạm vi chính để đầu tư phát triển NGS là khả năng ứng dụng rộng rãi của nó trong cả môi trường lâm sàng và nghiên cứu.
Trong môi trường lâm sàng, NGS được sử dụng để chẩn đoán các rối loạn khác nhau, thông qua việc xác định các đột biến dòng mầm hoặc soma. Sự chuyển đổi sang NGS trong thực hành lâm sàng được chứng minh bằng sức mạnh của kỹ thuật kết hợp với chi phí liên tục giảm.
NGS cũng là một công cụ có giá trị trong nghiên cứu metagenomics và được sử dụng để chẩn đoán, theo dõi và quản lý bệnh truyền nhiễm.
Vào năm 2020, các phương pháp NGS đóng vai trò quan trọng trong việc mô tả bộ gen của SARS-CoV-2 và không ngừng góp phần theo dõi đại dịch Covid 19.
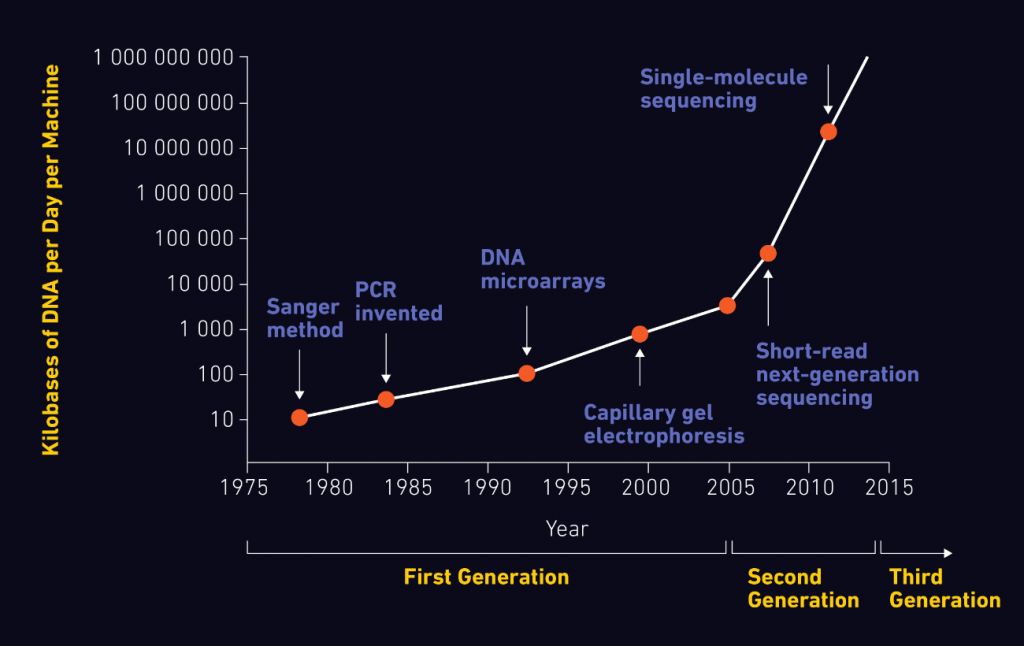
Hình 1: Sự phát triển của các công nghệ giải trình tự
Phương pháp giải trình tự thế hệ mới
Thuật ngữ NGS thường được hiểu là công nghệ 2G, tuy nhiên, kể từ đó, các công nghệ giải trình tự thế hệ thứ ba (3G) và thứ tư (4G) đã phát triển để hoạt động dựa trên các nguyên tắc cơ bản khác nhau.
Nền tảng giải trình tự và công nghệ giải trình tự
Các phương pháp giải trình tự thế hệ thứ hai được thiết lập tốt và có nhiều điểm chung.
Tuy nhiên, chúng có thể được chia nhỏ theo các cơ chế hóa học phát hiện cơ bản bao gồm giải trình tự bằng cách thắt (kết hợp bóng nano) và giải trình tự bằng tổng hợp (SBS), phân chia tiếp thành phát hiện proton, giải trình tự pyro và kết thúc thuận nghịch (Hình 2).

Hình 2: Các nền tảng giải trình tự 2G
Giải trình tự thế hệ 2
Trình tự phát hiện proton dựa vào việc đếm các ion hydro được giải phóng trong quá trình trùng hợp ADN. Không giống như các kỹ thuật khác, công nghệ này không sử dụng huỳnh quang và không sử dụng các nucleotide hoặc quang học đã được biến đổi. Thay vào đó, sự thay đổi độ pH được phát hiện bởi chip cảm biến bán dẫn và chuyển đổi thành thông tin kỹ thuật số.
Phương pháp pyrophosphate sử dụng phương pháp phát hiện sự tạo thành pyrophosphate và giải phóng ánh sáng để xác định xem liệu một bazơ cụ thể có được kết hợp trong chuỗi ADN hay không.
Cho đến nay, phương pháp SBS phổ biến nhất là giải trình tự kết thúc thuận nghịch sử dụng ”khuếch đại cầu”. Trong quá trình phản ứng tổng hợp, các mảnh liên kết với oligonucleotide trên tế bào dòng chảy, tạo ra một cầu nối từ một phía của trình tự (P5 oligo trên tế bào dòng chảy) đến bên kia (P7), sau đó được khuếch đại. Các nucleotide có nhãn huỳnh quang bổ sung được phát hiện bằng cách sử dụng hình ảnh trực tiếp.
Không giống như SBS, giải trình tự bằng cách thắt không sử dụng DNA polymerase để tạo chuỗi thứ hai. Thay vào đó, độ nhạy của DNA ligase đối với các cặp bazơ không khớp được sử dụng, với huỳnh quang được tạo ra được sử dụng để xác định trình tự đích. Hình ảnh kỹ thuật số được chụp sau mỗi phản ứng sẽ được sử dụng để phân tích.
Giải trình tự quả cầu nano ADN là một hình thức giải trình tự bằng cách thắt để khai thác sự sao chép vòng tròn lăn. Các bản sao ADN được nối được nén thành các quả cầu nano ADN và liên kết với các slide giải trình tự trong một mạng lưới dày đặc các điểm sẵn sàng cho các phản ứng giải trình tự dựa trên phương pháp thắt. Trong khi kỹ thuật bóng nano giúp giảm chi phí vận hành thì các chuỗi ngắn được tạo ra có thể gây khó khăn cho việc đọc bản đồ.
Công nghệ 2G NGS nói chung mang lại một số lợi thế so với các kỹ thuật giải trình tự thay thế, bao gồm khả năng tạo ra các lần đọc trình tự một cách nhanh chóng, nhạy cảm và tiết kiệm chi phí.
Tuy nhiên, cũng có những nhược điểm, bao gồm việc giải thích kém về homopolyme và sự kết hợp các dNTP không chính xác của polymerase với nhau, dẫn đến lỗi giải trình tự.
Độ dài đọc ngắn cũng tạo ra nhu cầu bao phủ trình tự sâu hơn để cho phép lắp ráp bộ gen cuối cùng và đường viền chính xác.
Nhược điểm chính của tất cả các kỹ thuật 2G NGS là cần khuếch đại PCR trước khi giải trình tự. Điều này có liên quan đến sai lệch PCR trong quá trình chuẩn bị thư viện (nội dung GC trình tự, độ dài đoạn và phân tập sai) và phân tích (lỗi cơ sở/ưu tiên các trình tự nhất định hơn các trình tự khác).
Giải trình tự thế hệ 3
Sự ra đời của công nghệ giải trình tự 3G giúp loại bỏ nhu cầu PCR, giải trình tự các phân tử đơn lẻ mà không cần các bước khuếch đại trước đó.
Công nghệ giải trình tự phân tử đơn (SMS) đầu tiên được phát triển bởi Stephen Quake và các đồng nghiệp. Ở đây, thông tin trình tự thu được bằng cách sử dụng DNA polymerase bằng cách theo dõi sự kết hợp của các nucleotide được đánh dấu huỳnh quang với các chuỗi ADN với độ phân giải bazơ duy nhất. Tùy thuộc vào phương pháp và công cụ được sử dụng, một số ưu điểm của 3G NGS bao gồm:
- Giám sát thời gian thực của việc kết hợp nucleotide
- Trình tự không thiên vị
- Độ dài đọc dài hơn
Tuy nhiên, chi phí cao, tỷ lệ lỗi cao, số lượng lớn dữ liệu tuần tự và độ sâu đọc thấp có thể là vấn đề của 3G NGS.
Giải trình tự thế hệ 4
Trong hệ thống 4G, trình tự phân tử đơn của 3G được kết hợp với công nghệ giải trình tự DNA nanopore
Tương tự như 3G, công nghệ nanopore không yêu cầu khuếch đại và sử dụng khái niệm giải trình tự phân tử đơn lẻ nhưng với sự tích hợp của các lỗ sinh học nhỏ có đường kính nano (nanopores) mà qua đó phân tử đơn lẻ đi qua và được xác định.
Các hệ thống 4G hiện cung cấp khả năng quét toàn bộ chuỗi gen nhanh nhất nhưng vẫn khá đắt, dễ xảy ra lỗi so với các kỹ thuật 2G và còn tương đối mới. Do đó, hiện tại có ít dữ liệu rộng rãi hơn cho kỹ thuật này.
Các bước chính của phương pháp giải trình tự 2G và chuẩn bị thư viện giải trình tự thế hệ mới
Bất kể phương pháp 2G NGS được chọn là gì, có một số bước chính phải được điều chỉnh và tối ưu hóa cho mục tiêu (ARN hoặc ADN) và hệ thống giải trình tự đã chọn.
(1) Chuẩn bị mẫu (tiền xử lý)
Axit nucleic (ADN hoặc ARN) được chiết xuất từ các mẫu sinh phẩm đã chọn (máu, đờm, tủy xương…).
Các mẫu chiết được kiểm tra kiểm soát chất lượng (QC), sử dụng các phương pháp tiêu chuẩn (đo quang phổ, đo huỳnh quang hoặc điện di trên gel). Nếu sử dụng ARN, điều này phải được sao chép ngược thành cDNA, tuy nhiên một số bộ công cụ chuẩn bị thư viện có thể bao gồm bước này.
(2) Chuẩn bị thư viện
Sự phân mảnh ngẫu nhiên của cDNA hoặc ADN, thường được thực hiện bằng cách xử lý enzyme hoặc siêu âm.
Độ dài đoạn tối ưu phụ thuộc vào nền tảng đang được sử dụng. Có thể cần phải chạy một lượng nhỏ mẫu đã phân mảnh trên gel điện di khi tối ưu hóa quá trình này.
Những đoạn này sau đó được sửa chữa cuối cùng và nối thành các đoạn ADN chung nhỏ hơn gọi là bộ điều hợp. Bộ điều hợp có độ dài xác định với các trình tự oligome đã biết để tương thích với nền tảng giải trình tự được áp dụng và có thể xác định được nơi thực hiện giải trình tự ghép kênh. Trình tự ghép kênh, bằng cách sử dụng các trình tự bộ điều hợp riêng lẻ cho mỗi mẫu, cho phép một số lượng lớn thư viện được gộp lại và sắp xếp theo trình tự đồng thời trong một lần chạy. Nhóm các đoạn DNA có gắn bộ điều hợp này được gọi là thư viện giải trình tự.
Sau đó, việc lựa chọn kích thước có thể được thực hiện bằng điện di trên gel hoặc sử dụng hạt từ tính để loại bỏ bất kỳ đoạn nào quá ngắn hoặc quá dài để đạt hiệu suất tối ưu trên nền tảng giải trình tự và giao thức đã chọn. Sau đó, việc làm giàu và khuếch đại thư viện được thực hiện bằng cách sử dụng PCR.
Trong các kỹ thuật liên quan đến PCR nhũ tương, mỗi đoạn được liên kết với một hạt nhũ tương duy nhất, hạt này sẽ tiếp tục tạo thành cơ sở cho các cụm giải trình tự. Quá trình khuếch đại thường được theo sau bởi bước “làm sạch” (ví dụ: sử dụng hạt từ tính) để loại bỏ các đoạn không mong muốn và cải thiện hiệu quả giải trình tự.
Các thư viện cuối cùng có thể trải qua quá trình kiểm tra QC bằng qPCRđể xác nhận chất lượng và số lượng ADN. Điều này cũng sẽ cho phép chuẩn bị nồng độ mẫu chính xác để giải trình tự.
(3) Giải trình tự
Tùy thuộc vào nền tảng và hóa học đã chọn, quá trình khuếch đại dòng vô tính của các đoạn thư viện có thể xảy ra trước khi nạp trình tự sắp xếp (PCR nhũ tương) hoặc trên chính trình tự sắp xếp (PCR cầu nối). Trình tự sau đó được phát hiện và báo cáo theo nền tảng đã chọn.
(4) Phân tích dữ liệu
Các tệp dữ liệu được tạo sẽ được phân tích tùy thuộc vào quy trình làm việc được sử dụng. Phương pháp phân tích phụ thuộc nhiều vào mục đích nghiên cứu.
Mặc dù chúng có thể làm giảm số lượng mẫu có thể được phân tích trong một lần chạy nhất định, nhưng trình tự cặp đầu cuối và cặp đôi mang lại lợi thế trong phân tích dữ liệu xuôi dòng, đặc biệt đối với các tổ hợp de novo. Các kỹ thuật này liên kết các lần đọc trình tự với nhau được đọc từ cả hai đầu của một đoạn (kết thúc theo cặp) hoặc được phân tách bằng một vùng ADN can thiệp (mate pair).
Rõ ràng có nhiều lựa chọn khi lựa chọn chiến lược giải trình tự. Sau đây là một số cân nhắc chính khi quyết định nền tảng chuẩn bị và sắp xếp thư viện thích hợp:
(a) Câu hỏi nghiên cứu đang được đặt ra
(b) Loại mẫu
(c) Trình tự đọc ngắn hoặc đọc dài
(d) Trình tự ADN hoặc ARN – bạn có cần xem bộ gen hoặc bản phiên mã không?
(e) Cần có toàn bộ bộ gen hay chỉ các vùng cụ thể?
(f) Cần có độ sâu đọc (phạm vi bao phủ) – dành riêng cho từng thử nghiệm
(g) Phương pháp chiết xuất
(h) Nồng độ mẫu
(i) Đọc một đầu, một đầu hoặc một cặp kết hợp
(j) Yêu cầu độ dài đọc cụ thể
(k) Các mẫu có thể được ghép kênh không?
(l) Công cụ tin sinh học – phụ thuộc vào thí nghiệm. Tùy thuộc vào mẫu và câu hỏi sinh học, toàn bộ quá trình phân tích trình tự có thể được điều chỉnh.
Giải trình tự đọc ngắn (short-read) và đọc dài (long-read)
Những ưu điểm và nhược điểm của giải trình tự đọc ngắn và đọc dài được tóm tắt trong Bảng 1 dưới đây.
Trình tự đọc ngắn:
- Độ chính xác trình tự cao hơn.
- Rẻ
- Có thể sắp xếp chuỗi ADN bị phân mảnh
Trình tự đọc dài:
- Có khả năng sắp xếp các vùng di truyền khó mô tả bằng trình tự đọc ngắn do trình tự lặp lại.
- Có khả năng giải quyết sự sắp xếp lại cấu trúc hoặc các vùng tương đồng
- Có thể đọc toàn bộ bản phiên mã ARN để xác định dạng đồng phân cụ thể
- Hỗ trợ lắp ráp bộ gen de novo
Bảng 1a: Ưu điểm của trình tự đọc ngắn và đọc dài
Trình tự đọc ngắn:
- Không thể giải quyết các biến thể cấu trúc, phân kỳ các alen hoặc phân biệt các vùng gen có tính tương đồng cao
- Không thể cung cấp vùng phủ sóng cho một số vùng lặp đi lặp lại
Trình tự đọc dài:
- Độ chính xác mỗi lần đọc thấp hơn
- Nhưng thách thức về tin sinh học, do sai lệch phạm vi bao phủ, tỷ lệ lỗi cao trong phân bổ cơ sở, khả năng mở rộng và tính sẵn có hạn chế của các đường ống thích hợp
Bảng 1b: Hạn chế của trình tự đọc ngắn và đọc dài
Sự khác biệt giữa giải trình tự toàn bộ exome và toàn bộ bộ gen
Giải trình tự toàn bộ bộ gen (Whole Genome Sequencing – WGS) là dạng NGS được sử dụng rộng rãi nhất và đề cập đến việc phân tích toàn bộ trình tự nucleotide của bộ gen.
Giải trình tự toàn bộ exome (Whole Exome Sequencing – WES) là một dạng giải trình tự có mục tiêu, chỉ giải quyết các exon mã hóa protein.
Ở người, các trình tự exome chỉ chiếm khoảng 2% bộ gen và do đó mang đến cơ hội nghiên cứu sâu hơn ở những khu vực này.
Do giảm bớt gánh nặng trình tự, WES cũng có thể cung cấp tùy chọn tiết kiệm chi phí hơn WGS và giảm khối lượng cũng như độ phức tạp của dữ liệu trình tự tổng hợp.
Tuy nhiên, bằng cách chỉ giải trình tự một phần của bộ gen, thông tin quan trọng có thể bị bỏ sót và cơ hội khám phá mới sẽ giảm đi. Mặc dù chi phí tăng lên, mặc dù giảm nhanh chóng và những thách thức phân tích dữ liệu liên quan, WGS do đó cung cấp một phân tích mạnh mẽ hơn có thể tiết lộ một bức tranh hoàn chỉnh hơn.
Phân tích dữ liệu giải trình tự thế hệ mới
Bất kỳ loại công nghệ NGS nào cũng tạo ra một lượng dữ liệu đầu ra đáng kể.
Khái niệm cơ bản về phân tích trình tự tuân theo quy trình làm việc tập trung bao gồm bước QC đọc thô, xử lý trước và ánh xạ, sau đó là xử lý sau căn chỉnh, chú thích biến thể, gọi biến thể và trực quan hóa.
Việc đánh giá dữ liệu trình tự thô là bắt buộc để xác định chất lượng của chúng và mở đường cho tất cả các phân tích tiếp theo. Quá trình này có thể cung cấp cái nhìn tổng quát về số lượng và độ dài của các lần đọc, bất kỳ trình tự gây ô nhiễm nào hoặc bất kỳ lần đọc nào có phạm vi bao phủ thấp.
Một trong những ứng dụng được thiết lập tốt nhất để tính toán số liệu thống kê kiểm soát chất lượng của các lần đọc trình tự là FastQC.
Tuy nhiên, để xử lý trước thêm, chẳng hạn như lọc đọc và cắt xén, cần có các công cụ bổ sung. Việc cắt bớt các cơ sở ở cuối quá trình đọc và loại bỏ các chuỗi bộ điều hợp còn sót lại thường cải thiện chất lượng dữ liệu.
Gần đây hơn, các công cụ cực nhanh đã được giới thiệu, chẳng hạn như fastp, có thể thực hiện kiểm soát chất lượng, lọc đọc và hiệu chỉnh cơ sở trên dữ liệu tuần tự, kết hợp hầu hết các tính năng từ các ứng dụng truyền thống đồng thời chạy nhanh hơn hai đến năm lần so với bất kỳ ứng dụng nào trong số đó.
Sau khi chất lượng của các lần đọc đã được kiểm tra và quá trình xử lý trước được thực hiện, bước tiếp theo sẽ phụ thuộc vào sự tồn tại của bộ gen tham chiếu (reference genome). Trong trường hợp tập hợp bộ gen de novo, các trình tự được tạo ra sẽ được sắp xếp thành các đường viền bằng cách sử dụng các vùng chồng chéo của chúng. Điều này thường được thực hiện với sự hỗ trợ của quy trình xử lý có thể bao gồm các bước giàn giáo để giúp sắp xếp thứ tự, định hướng và loại bỏ các vùng lặp đi lặp lại, do đó làm tăng tính liên tục của quá trình lắp ráp.
Nếu các chuỗi được tạo được ánh xạ ( căn chỉnh) với bộ gen hoặc bản phiên mã tham chiếu, thì có thể xác định được các biến thể so với trình tự tham chiếu.
Ngày nay, có rất nhiều công cụ lập bản đồ (mapping tools), đã được điều chỉnh để xử lý lượng dữ liệu ngày càng tăng do NGS tạo ra, khai thác các tiến bộ công nghệ và giải quyết sự phát triển giao thức.
Một khó khăn do số lượng người lập bản đồ ngày càng tăng là có thể tìm được bản đồ phù hợp nhất. Thông tin thường được rải rác qua các ấn phẩm, mã nguồn (nếu có), sách hướng dẫn và các tài liệu khác.
Một số công cụ cũng sẽ cung cấp tính năng kiểm tra chất lượng ánh xạ cần thiết vì một số thành kiến sẽ chỉ hiển thị sau bước lập bản đồ.
Tương tự như kiểm soát chất lượng trước khi lập bản đồ, việc xử lý chính xác các lần đọc bản đồ là một bước quan trọng, trong đó các lần đọc bản đồ trùng lặp (bao gồm nhưng không giới hạn ở các tạo phẩm PCR) sẽ bị loại bỏ.
Đây là một phương pháp được tiêu chuẩn hóa và hầu hết các công cụ đều có chung các tính năng.
Khi các lần đọc đã được ánh xạ và xử lý, chúng cần được phân tích theo kiểu thử nghiệm cụ thể, cái được gọi là phân tích biến thể. Bước này có thể xác định các đa hình nucleotide đơn (SNP), RNA-seq và nhiều hơn nữa.
Mặc dù có vô số công cụ để lắp ráp, căn chỉnh và phân tích bộ gen, nhưng vẫn luôn cần có các phiên bản mới và cải tiến để đảm bảo rằng độ nhạy, độ chính xác và độ phân giải có thể phù hợp với các kỹ thuật NGS tiến bộ nhanh chóng.
Bước cuối cùng là trực quan hóa, độ phức tạp của dữ liệu có thể đặt ra một thách thức đáng kể. Tùy thuộc vào thí nghiệm và câu hỏi nghiên cứu được đặt ra, có thể sử dụng một số công cụ.
Nếu có sẵn bộ gen tham chiếu thì Trình xem bộ gen tích hợp (Integrative Genomics Viewer -IGV) là một lựa chọn phổ biến, cũng như Trình duyệt bộ gen. (Genome Browser) Nếu các thử nghiệm bao gồm WGS hoặc WES thì Variant Explorer là một công cụ đặc biệt tốt vì nó có thể được sử dụng để sàng lọc hàng nghìn biến thể và cho phép người dùng tập trung vào những phát hiện quan trọng nhất của họ.
Các công cụ trực quan như VISTA cho phép so sánh giữa các trình tự gen khác nhau.
Các chương trình phù hợp với tập hợp bộ gen de novo bị hạn chế hơn. Tuy nhiên, các công cụ như Bandage Icarus đã được sử dụng để khám phá và phân tích bộ gen được tập hợp.
Hạn chế của công nghệ giải trình tự thế hệ mới
NGS đã cho phép chúng tôi khám phá và nghiên cứu bộ gen theo những cách chưa từng có trước đây. Tuy nhiên, sự phức tạp của quá trình xử lý mẫu NGS đã bộc lộ những điểm nghẽn trong việc quản lý, phân tích và lưu trữ bộ dữ liệu.
Một trong những thách thức chính là tài nguyên tính toán cần thiết cho việc lắp ráp, chú thích và phân tích dữ liệu giải trình tự.
Lượng dữ liệu khổng lồ được tạo ra từ phân tích NGS là một thách thức quan trọng khác.

Các trung tâm dữ liệu đang đạt mức dung lượng lưu trữ cao và không ngừng cố gắng đáp ứng nhu cầu ngày càng tăng, có nguy cơ mất dữ liệu vĩnh viễn.
Nhiều chiến lược khác liên tục được đề xuất nhằm mục đích tăng hiệu quả, giảm lỗi trình tự, tối đa hóa khả năng tái tạo và đảm bảo quản lý dữ liệu chính xác.
Ứng dụng công nghệ giải trình tự thế hệ mới
Kể từ đầu những năm 2000, giải trình tự thế hệ mới đã trở thành một công cụ vô giá trong cả môi trường nghiên cứu và lâm sàng/ chẩn đoán cho y học hiện đại và khám phá thuốc, với việc sử dụng các phương pháp bao gồm WGS, WES, giải trình tự mục tiêu (targeted sequencing), giải trình tự phiên mã (transcriptome), giải trình tự biểu sinh (epigenome) và giải trình tự metagenome ngày càng tăng đáng kể. Hình 3 dưới đây sẽ tóm tắt các quy trình công việc và các tùy chọn để nhắm mục tiêu các bộ dữ liệu khác nhau.

Hình 3: Các chiến lược giải trình tự khả thi cho các loại mẫu khác nhau.
Thông qua WGS, các nhà nghiên cứu có thể nghiên cứu không chỉ các gen và sự liên quan của chúng đối với bệnh tật ở người và động vật mà còn cả các đặc điểm của quần thể vi sinh vật và nông nghiệp, cung cấp dữ liệu dịch tễ học và tiến hóa quan trọng.
Cho đến nay, đã có rất nhiều nghiên cứu trong đó các đột biến, sắp xếp lại và các sự kiện hợp nhất được xác định bằng cách sử dụng WGS. Hiện tại, WGS được sử dụng để giám sát tình trạng kháng kháng sinh, một trong những thách thức lớn về sức khỏe toàn cầu.
Do chi phí không ngừng giảm nên WGS được sử dụng thường xuyên hơn để sắp xếp lại toàn bộ bộ gen người trong các mẫu lâm sàng và có thể sớm trở thành thói quen trong thực hành lâm sàng.
Cuối cùng, WGS sẽ cần thiết để gán chức năng cho phần lớn còn lại của bộ gen và giải mã vai trò của nó đối với các bệnh tật.
Bản chất tập trung hơn của chúng làm cho WES và giải trình tự mục tiêu trở thành những lựa chọn hấp dẫn cho các nghiên cứu lâm sàng và dân số. Mặc dù có nhiều hạn chế hơn như tên gọi, WES là một công cụ lâm sàng quan trọng trong lĩnh vực y học cá nhân hóa.
Chẩn đoán di truyền đối với một số bệnh nhất định, như ung thư, cũng như đặc tính di truyền đối với các rối loạn khác có thể đạt được bằng phương pháp này theo cách tiết kiệm chi phí hơn so với WGS.
Ngoài nhiều ứng dụng mà NGS có trong việc giải trình tự ADN, giải trình tự thế hệ mới còn có thể được sử dụng để phân tích ARN (RNA-seq).
Điều này cho phép xác định bộ gen của virus ARN, chẳng hạn như SARS và cúm.
Điều quan trọng là RNA-seq thường được sử dụng trong các nghiên cứu định lượng, không chỉ tạo điều kiện thuận lợi cho việc xác định các gen phiên mã trong bộ gen ADN mà còn cả mức độ chúng được phiên mã (mức độ phiên mã) tùy theo mức độ phong phú tương đối của các bản phiên mã ARN.
Khả năng sắp xếp lại các trình tự ADN cũng có thể được xác định thông qua việc xác định các bản phiên mã mới.
Giải trình tự biểu sinh (Epigenomic sequencing) cho phép nghiên cứu những thay đổi gây ra bởi sửa đổi histone và quá trình methyl hóa ADN.
Có nhiều phương pháp khác nhau được sử dụng để nghiên cứu các cơ chế biểu sinh, bao gồm giải trình tự bisulfate toàn bộ bộ gen (WGBS), kích thích miễn dịch nhiễm sắc thể (ChIP-seq) và kích thích miễn dịch phụ thuộc methyl hóa (MeDIP-seq), sau đó là giải trình tự. Tùy thuộc vào phương pháp đã chọn, hồ sơ chỉnh sửa methylome và histone DNA hoàn chỉnh có thể được lập bản đồ và nghiên cứu, thu được những hiểu biết sâu sắc về cơ chế điều hòa bộ gen.
Giải trình tự metagenomic (Metagenomic sequencing) có thể cung cấp thông tin cho các mẫu được thu thập trong một môi trường cụ thể. Phương pháp này cho phép so sánh sự khác biệt và tương tác giữa các quần thể vi sinh vật hỗn hợp, cũng như phản ứng của vật chủ.
Một số ứng dụng tiềm năng của giải trình tự metagenomics bao gồm nhưng không giới hạn ở chẩn đoán bệnh truyền nhiễm và giám sát nhiễm trùng, theo dõi tình trạng kháng kháng sinh, nghiên cứu hệ vi sinh vật và phát hiện mầm bệnh.
Những tiến bộ công nghệ trong việc chuẩn bị mẫu, công nghệ giải trình tự và phân tích dữ liệu có nghĩa là NGS cũng đang được sử dụng ở cấp độ tế bào đơn lẻ (single cell) để nghiên cứu tính không đồng nhất và những thay đổi hiếm gặp trong ADN, ARN và biểu sinh
Tài liệu tham khảo
- Watson JD, Crick FHC. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737-738. doi:10.1038/171737a0
- Franklin RE, Gosling RG. Molecular configuration in sodium thymonucleate. Nature. 1953;171(4356):740-741. doi:10.1038/171740a0
- Holley RW, Apgar J, Everett GA, et al. Structure of a ribonucleic acid. Science (80- ). 1965;147(3664):1462-1465. doi:10.1126/science.147.3664.1462
- Fiers W, Contreras R, Duerinck F, et al. Complete nucleotide sequence of bacteriophage MS2 RNA: Primary and secondary structure of the replicase gene. Nature. 1976;260(5551):500-507. doi:10.1038/260500a0
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463-5467. doi:10.1073/pnas.74.12.5463
- Smith LM, Sanders JZ, Kaiser RJ, et al. Fluorescence detection in automated DNA sequence analysis. Nature. 1986;321(6071):674-679. doi:10.1038/321674a0
- Hood LE, Hunkapiller MW, Smith LM. Automated DNA sequencing and analysis of the human genome. Genomics. 1987;1(3):201-212. doi:10.1016/0888-7543(87)90046-2
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387-402. doi:10.1146/annurev.genom.9.081307.164359
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135-1145. doi:10.1038/nbt1486
- Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet. 2019;10(MAY):426. doi:10.3389/fgene.2019.00426
- Lefterova MI, Suarez CJ, Banaei N, Pinsky BA. Next-Generation Sequencing for Infectious Disease Diagnosis and Management: A Report of the Association for Molecular Pathology. J Mol Diagnostics. 2015;17(6):623-634
- Mostafa HH, Fissel JA, Fanelli B, et al. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect covid-19 patients. MBio. 2020;11(6):1-13. doi:10.1128/mBio.01969-20
- Ronaghi M, Karamohamed S, Pettersson B, Uhlén M, Nyrén P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 1996;242(1):84-89. doi:10.1006/abio.1996.0432
- Slatko BE, Gardner AF, Ausubel FM. Overview of Next-Generation Sequencing Technologies. Curr Protoc Mol Biol. 2018;122(1):e59. doi:10.1002/cpmb.59
- Buermans HPJ, den Dunnen JT. Next generation sequencing technology: Advances and applications. Biochim Biophys Acta – Mol Basis Dis. 2014;1842(10):1932-1941. doi:10.1016/j.bbadis.2014.06.015
- Porreca GJ. Genome sequencing on nanoballs. Nat Biotechnol. 2010;28(1):43-44. doi:10.1038/nbt0110-43
- Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012. doi:10.1155/2012/251364
- Timp W, Mirsaidov UM, Wang D, Comer J, Aksimentiev A, Timp G. Nanopore sequencing: Electrical measurements of the code of life. IEEE Trans Nanotechnol. 2010;9(3):281-294. doi:10.1109/TNANO.2010.2044418
- Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14(6):405. doi:10.1186/gb-2013-14-6-405
- Hess JF, Kohl TA, Kotrová M, et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol Adv. 2020;41. doi:10.1016/j.biotechadv.2020.107537
- Wu J, Wu M, Chen T, Jiang R. Whole genome sequencing and its applications in medical genetics. Quant Biol. 2016;4(2):115-128. doi:10.1007/s40484-016-0067-0
- Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27(9):522-530. doi:10.1016/j.tibtech.2009.05.006
- Kwong JC, Mccallum N, Sintchenko V, Howden BP. Whole genome sequencing in clinical and public health microbiology. Pathology. 2015;47(3):199-210. doi:10.1097/PAT.0000000000000235
- Ozsolak F, Milos PM. RNA sequencing: Advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87-98. doi:10.1038/nrg2934
- Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57-63. doi:10.1038/nrg2484
- Xing X, Zhang B, Li D, Wang T. Comprehensive whole DNA methylome analysis by integrating MeDIP-seq and MRE-seq. In: Methods in Molecular Biology. Vol 1708. Humana Press Inc.; 2018:209-246. doi:10.1007/978-1-4939-7481-8_12
- Ku CS, Naidoo N, Wu M, Soong R. Studying the epigenome using next generation sequencing. J Med Genet. 2011;48(11):721-730. doi:10.1136/jmedgenet-2011-100242
- Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341-355. doi:10.1038/s41576-019-0113-7




